这篇文章主要对微生物组学研究中常出现的概念进行整理,
资料摘抄原文如下:
- 细菌16s rDNA微生物多样性研究中可变区的选择
- 16S 基础知识、分析工具和分析流程详解
- 16S/18S/ITS 扩增子测序
- 微生物组学数据分析工具综述 | 16S+宏基因组+宏病毒组+宏转录组–转载
微生物组学的常见分类
建立在高通量测序基础上的微生物群落研究,当前主要有三大类:
- 基于16S/18S/ITS等扩增子做物种分类的Metataxanomics、
- 鸟枪法打断全基因组DNA序列的Metagenomics
- 和基于mRNA信息的宏转录组方法Meta-transcriptomics。

###16S/18S/ITS扩增子测序
16S rDNA是细菌分类学研究中最常用的“分子钟”,其序列包含9个可变区(Variable region)和10个保守区(constant region)。可变区因细菌而异,且变异程度与细菌的系统发育密切相关。通过检测16S rDNA的序列变异和丰度,可以了解环境样品中群落多样性信息。基于16S rDNA的分析在微生物分类鉴定、微生态研究等方面起到重要作用。
16S rDNA结构:16s rRNA基因的序列(约1500bp)有10个保守区和9个高变区(v1-v9:长度分布范围约30~100bp)之分;保守区为所有细菌共有,细菌间无差别,能反映生物物种的亲缘关系,可变区具有属或种的特异性,序列则随菌间的亲缘关系不同而有一定的差异,所以能揭示生物物种的特征核酸序列,被认为是最适于细菌系统发育和分类鉴定的指标。根据保守区设计引物位点,扩增可变区获得的序列可以用于菌种鉴定。一种快速、廉价的菌种鉴定方法。
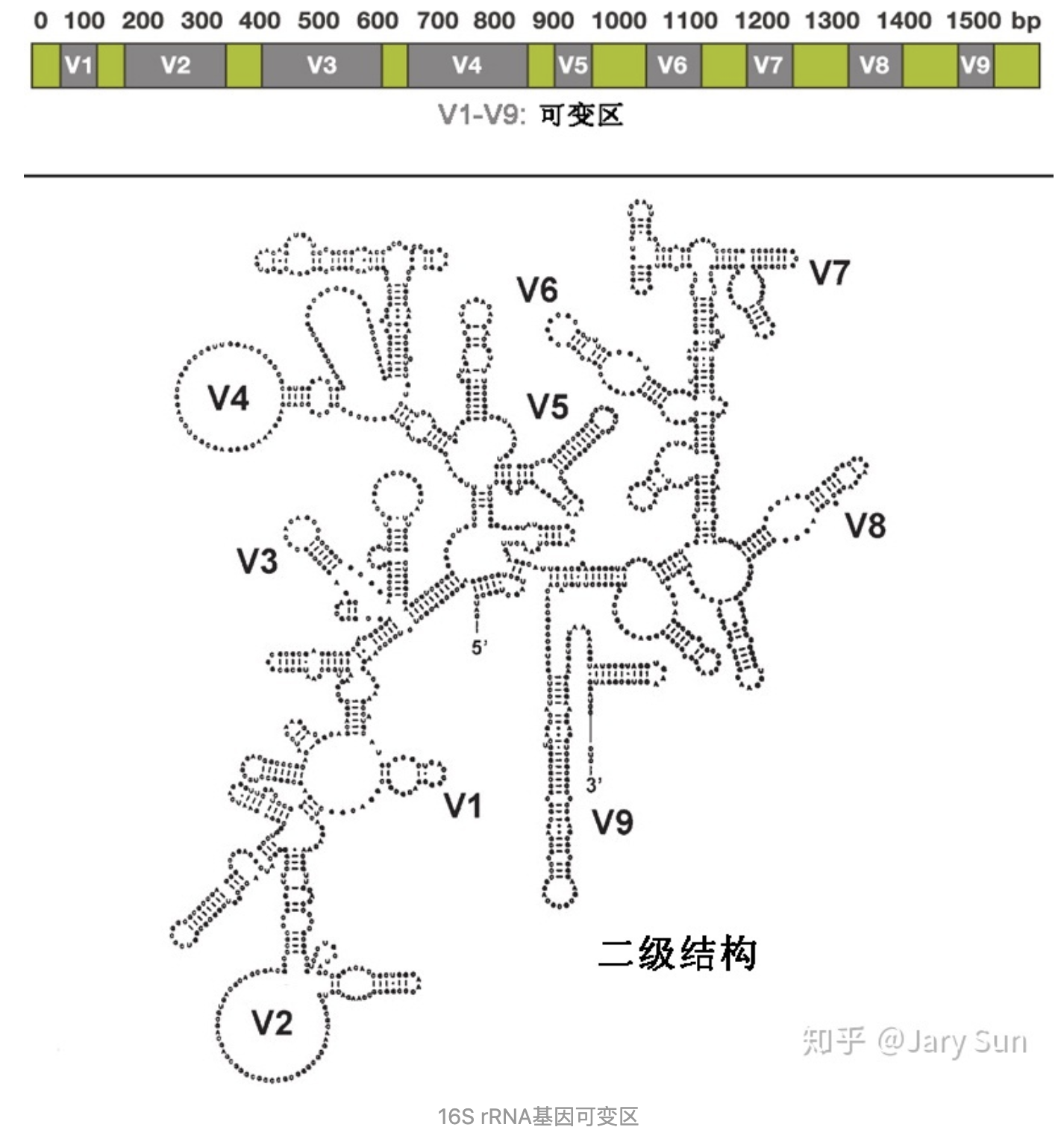
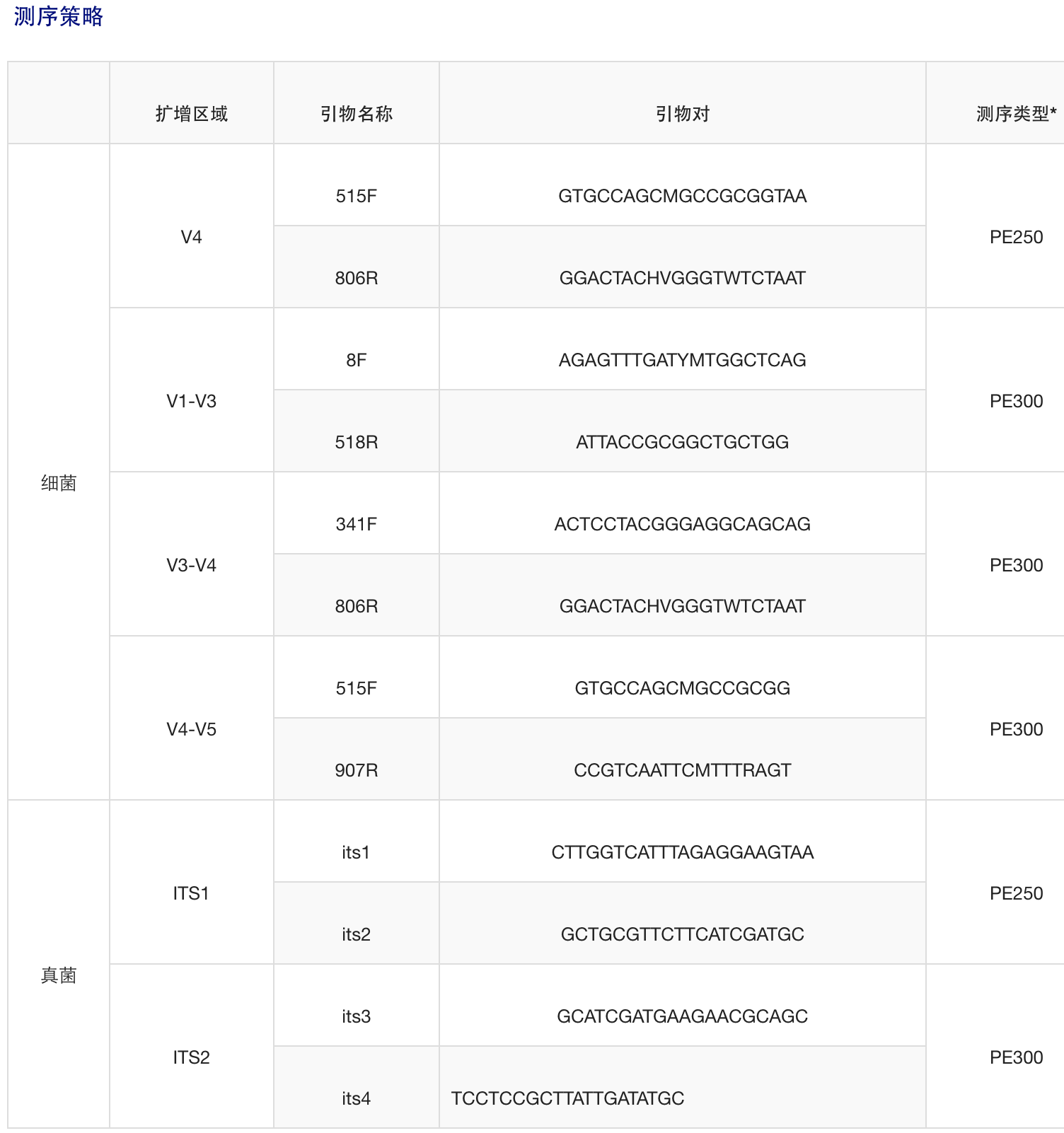
16s rRNA基因可变区考虑因素:
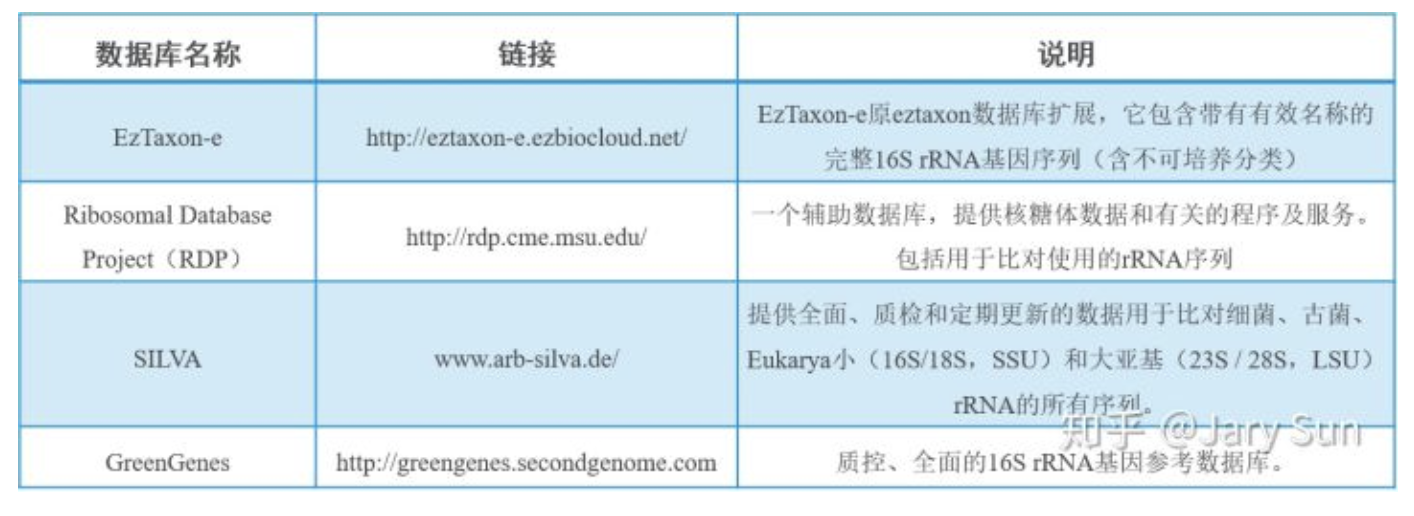
16s rRNA基因数据库
SILVA; GreenGenes, RDP, EzTaxon-e
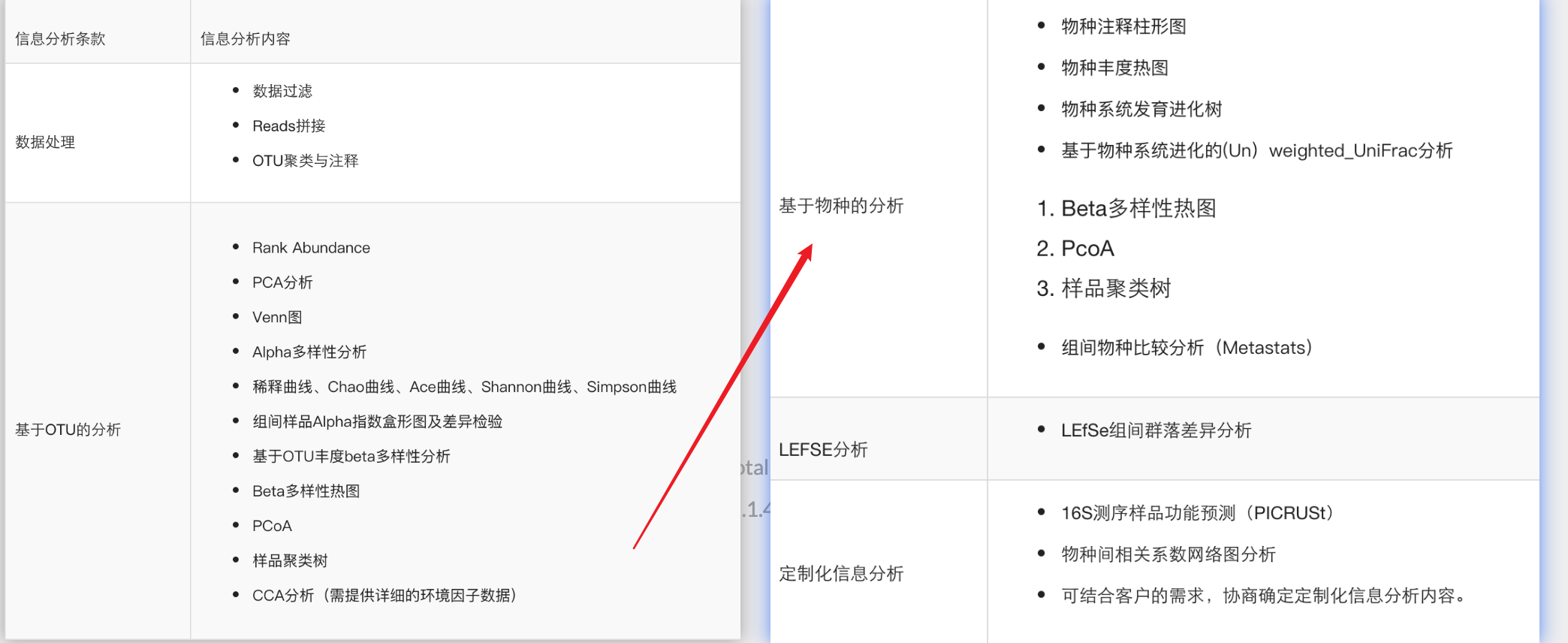
几个概念:
核糖体:Ribosome,由 RNA(rRNA)和蛋白质 组成,配合 tRNA 来翻译 mRNA。核糖体按沉降系数来分类,S就是沉降系数,原核70S,真核80S。我们一般研究微生物,70S,由50S和30S两个亚基组成。再细分为 5S、16S、23S,我们的 16S 就是指核糖体的亚基的一个组分,16S rRNA。(记住,这是原核生物核糖体的一个组分)
16S rRNA:这并不是我们的研究对象,因为我们测序的不是它,而是它对应在DNA双链上的基因序列,
16S rDNA。可以这样理解,我们所说的16S 就是指 16S rDNA。
分子钟:即氨基酸在单位时间以同样的速度进行置换。16S 的进化具有良好的时钟性质,在结构与功能上具有高度的保守性,在大多数原核生物中rDNA都具有多个拷贝,5S、16S、23S rDNA的拷贝数相同。16S rDNA由于大小适中,约1.5Kb左右,既能体现不同菌属之间的差异,又能利用测序技术较容易地得到其序列,故被细菌学家和分类学家接受。(来源百度)
OTU:即Operational Taxonomic Units的缩写(千万表手滑写成OUT,否则就OUT了),在系统发生学或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,属,种、分组等)设置的同一标志。理论上一个OTU代表一个微生物物种。
通过测序获得的大量reads,如何才能转变为我们需要的物种信息呢?首先需要对这些reads进行归类(cluster),通常在97%的相似水平划分为不同的OTU,将OTU代表序列与相应的微生物数据库比对(Silva、RDP、Greengene等),得到每个样本所含的物种信息,进而进行后续生物信息统计分析。
Alpha多样性:用于度量群落生态单样本的物种多样性,是反映丰富度和均匀度的综合指标。
菌群丰富度(Community richness)指数有:Chao、Ace,Chao或Ace指数越大,说明菌群丰富度越高。
菌群多样性(Community diversity)指数有:Shannon、Simpson,Shannon值越大,说明群落多样性越高;Simpson指数值越大,说明菌群多样性越低。
根据各样本生成的OTU,对样本序列进行随机取样,以取出的序列数及这些序列所能代表的OTU数构建曲线,计算样本的Alpha多样性。
Beta-diversity: Beta多样性用于不同生态系统之间多样性的比较,利用各样本序列间的进化关系及丰度信息来计算样本间距离,反映样本(组)间是否具有显著的微生物群落差异。目前应用比较多的是PCA、PCoA、NMDS分析等。由于微生物多样性研究通常会涉及到大样本数量的样本,因此通过Beta-diversity分析可以直观地反映样本组间的差异情况。距离越远,微生物群落差异越大,即相似性越高。
LEfSe
LEfSe分析即LDA Effect Size分析,多用于多个分组(≧2)之间的比较,或者进行亚组比较分析,进而找到组间在丰度上有显著差异的物种(biomaker)。基本过程是首先在多组样本中采用非参数因子Kruskal-Wallis秩和检验检测不同分组间丰度差异显著的物种;然后基于获得的显著差异物种,利用成组的Wilcoxon秩和检验进行组间差异分析;最后采用线性判别分析(LDA)对数据进行降维并评估差异显著的物种的影响力大小(即LDA score)。
18S rDNA或ITS(Internal Transcribed Spacer)被广泛应用在真菌分类鉴定中。18S rDNA在系统发育研究中较适用于种级以上阶元的分类;ITS属于中度保守区域,利用它可研究种及种以下的分类阶元。
局限性
16S虽然是一种相对快速和经济适用的方法,但是PCR导致了偏好的产生,这就降低了注释准确度。此外,由于原核、真核生物的“分类标签”完全不同,即使细菌和古菌的16S也相去甚远,以进化快著称的病毒更难以捕获。
常规分析流程
下机数据经过数据过滤,滤除低质量的reads,剩余高质量的Clean data方可用于后期分析;通过reads之间的Overlap关系将reads拼接成Tags;在给定的相似度下将Tags聚成OTU,然后通过OTU与数据库比对,对OTU进行物种注释;基于OTU和物种注释结果进行样品物种复杂度分析以及组间物种差异分析。
宏基因组
宏基因组有效避免了扩增偏差,由于是直接打断,理论上不限制物种(细菌、真菌、古菌、真核生物等,事实上当前宏基因组测序多还是以细菌为主),可能组装获得新基因乃至新物种信息,但根据取样情况可能存在少量或大量的宿主污染,因需组装,数据量要求大,成本贵、周期长。
宏基因组经典流程:环境微生物样本–Total DNA提取–文库构建–上机测序(经典短读长: illumina系列;长读长选择: PB, ONT)–数据质控(去除低质量和接头等,去除宿主基因组等干扰信息)–宏基因组组装–Contig Binning–基因组重建–分类注释(可基于reads、contig、bins、还原出来的基因组做物种注释)–其他下游分析。
宏转录组
宏转录组的好处是,跳出了DNA层面的束缚,可以获得实时活跃的、真正对群落有贡献的基因和通路,然而mRNA不如DNA稳定,此外多纯化和扩增的步骤也可能引入错误。
宏转录组也迎来了自己的专属软件–IMSA+A ( https://github.com/JeremyCoxBMI/IMSA-A )。IMSA+A在17年1月发表于Microbiome,是一种可应用于任意读长宏转录组学数据、可高效在同一份样品中鉴定出细菌、真菌、病毒的准确的分类分析的方法。