Nat. M.L. | 将多组学数据与图卷积网络整合以识别新的癌症基因及其相关的分子机制
题目:Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms
杂志:Nature Machine Intelligence
IF: 15
时间:12 April 2021
链接:https://www.nature.com/articles/s42256-021-00325-y
摘要
随着可用的高通量分子数据的增加,也为识别癌症基因带来了计算上的挑战。遗传和非遗传原因都有可能造成肿瘤的发生,这就需要开发预测模型来有效地整合不同的数据模式,同时具有可解释性。我们介绍了EMOGI,这是一种基于图卷积网络的可解释的机器学习方法,其通过结合多组学的泛癌症数据来预测癌症基因,如突变、拷贝数变化、DNA甲基化和基因表达,蛋白质-蛋白质相互作用(PPI) 网络等。在不同的PPI网络和数据集中,EMOGI总体来说比其他方法更准确。我们使用分层相关性传播,根据基因的分类是由相互作用组还是由任何一个全向性水平驱动,对基因进行分层,并确定PPI网络的重要模块。我们提出了165个新的癌症基因,这些基因不一定有反复的改变,但与已知的癌症基因有相互作用。而且我们发现,这些基因与功能缺失筛查中的基本基因相对应。我们相信,我们的方法可以为精准肿瘤学开辟新的途径,并可应用于预测癌症的生物标志物。
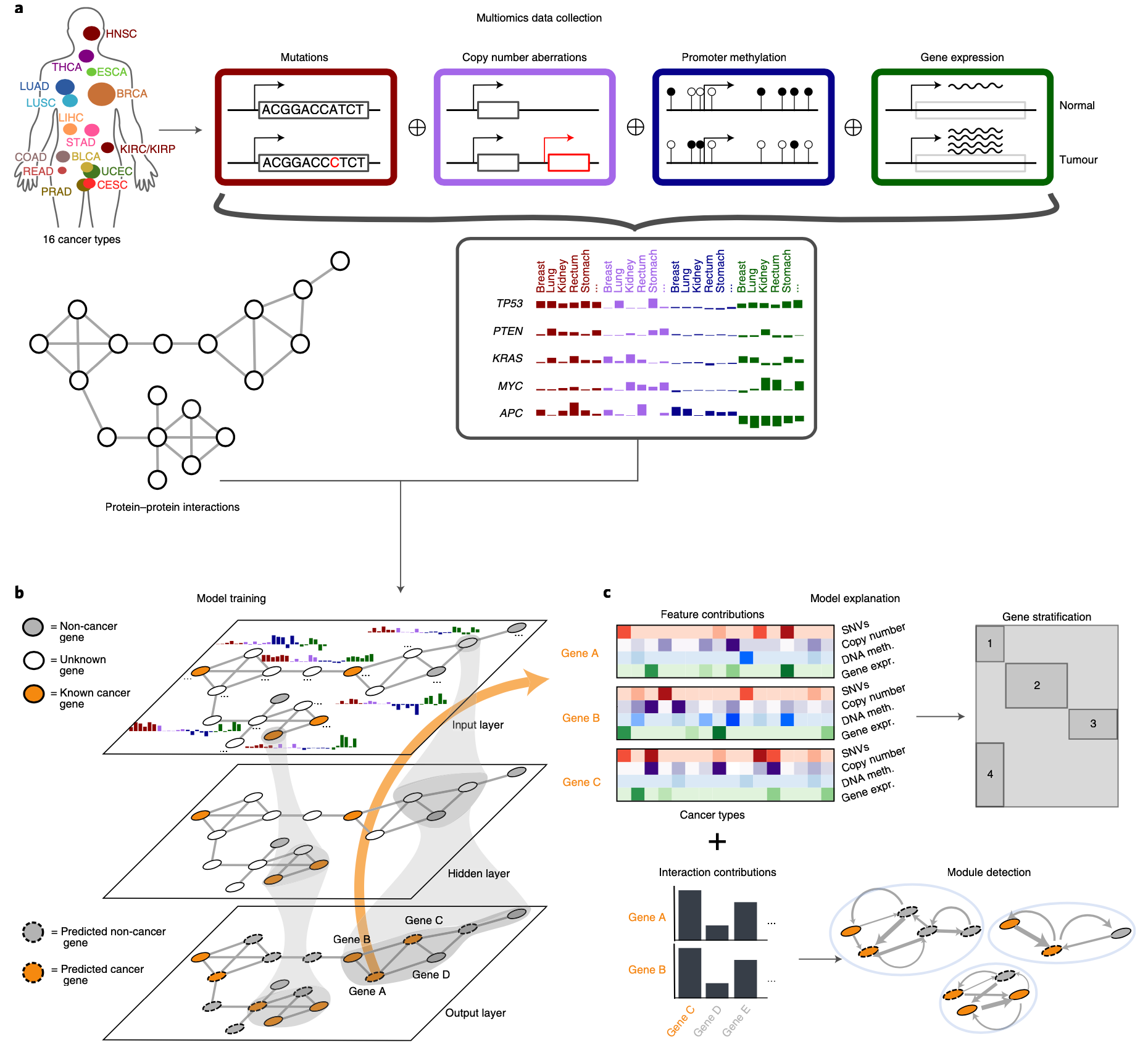
EMOGI框架的示意图。a, 数据收集和串联。计算16个TCGA肿瘤类型的所有基因的平均突变率、CNAs、DNA甲基化和基因表达变化,并在一个早期整合方案中进行连接。然后将得到的特征矩阵与PPI网络和一小部分高置信度的癌症/非癌症基因相结合,形成一个网络,其中节点对应于基因,边对应于它们之间已知的相互作用。每个节点/基因都有一个多维的特征向量(b,输入层)。
b,在EMOGI模型训练期间,特征通过连续的图卷积层进行转化(见方法),考虑到越来越大的邻域。输出层根据基因的输出概率将其分类为预测的癌症基因和非癌症基因。
c, 使用LRP(见方法)提取每个基因分类的最重要的特征(包括不同癌症类型的全能性水平和相互作用伙伴)。随后根据基因的特征贡献进行聚类,每个基因的相互作用贡献被用来检测癌症中具有重要基因-基因联系的模块。
Nat. M.L. | 用于估计抗SARS-CoV-2活动的机器学习平台
题目:A machine learning platform to estimate anti-SARS-CoV-2 activities
杂志:Nature Machine Intelligence
IF: 15
时间:13 May 2021
链接:https://www.nature.com/articles/s42256-021-00335-w
摘要
迫切需要针对COVID-19的药物发现和重新定位的策略。在此,我们提出了REDIAL-2020,一套用于估计小分子在一系列SARS-CoV-2相关检测中的活性的计算模型。模型是利用公开的高通量筛选数据,通过采用不同的描述符类型和各种机器学习策略来训练的。这里我们描述了11个模型的开发和使用,这些模型跨越了病毒进入、病毒复制、活病毒感染性、体外感染性和人类细胞毒性等领域。REDIAL-2020可通过DrugCentral网络门户(http://drugcentral.org/Redial)作为一个网络应用。该网络应用还提供相似性搜索结果,显示与查询最相似的分子,以及相关的实验数据。REDIAL-2020可以作为一个快速的在线工具,用于识别COVID-19治疗的活性分子。