文章信息
题目:Machine learning with random subspace ensembles identifies antimicrobial resistance determinants from pan-genomes of three pathogens
杂志:PLOS COMPUTATIONAL BIOLOGY
时间:March 2, 2020
链接: https://doi.org/10.1371/journal.pcbi.1007608
文章概述
抗菌剂耐药性的演变对全球公共卫生构成了持续的威胁。 测序工作已经获得了数千种耐药微生物分离物的基因组序列,需要强大的计算工具来系统地阐明AMR的遗传基础。
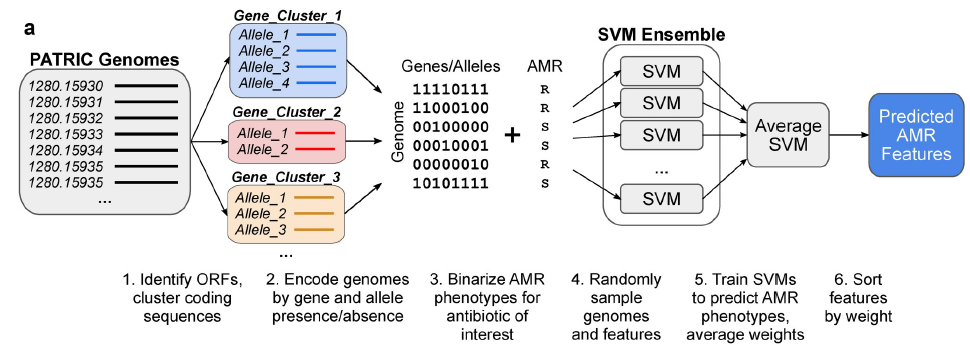
在这里,文章提出了一种可通用的机器学习工作流程,基于构建的参考菌株诊断泛基因组和训练随机子空间集合(RSEs),用于鉴定驱动AMR的遗传特征。这一工作流程对三种病原体的14种抗菌素的耐药性谱进行了研究,其中包括288种金黄色葡萄球菌、456种绿脓杆菌和1588种大肠杆菌基因组。文章发现,与常见的统计学检验和以前的集合方法相比,他们发现通过特征选择法检测已知的AMR更可靠,利用该方法共识别出45个已知的AMR基因和等位基因,以及25个由域级注释支持的候选关联。此外,发现来自于RSE方法的结果与现有的氟喹诺酮(FQ) 的理解是一致的。(FQ)抗药性是由于这三种生物体中主要药物靶点gyrA和parC的突变引起的,并表明这些基因在FQ抗药性方面的突变情况是简单的。随着更大的数据集的出现,我们希望这种方法能够更可靠地预测更多微生物病原体的AMR决定因素。
方法详解
数据来源:数据集是从PATRIC数据库下载的,共包括288,456和1588个金黄色葡萄球菌、铜绿假单胞菌和大肠杆菌的基因组数据。
数据预处理:鉴定开放阅读框,对编码基因进行聚类;由于AMR的致病变异通常存在于个体突变水平,他们对每个泛基因组的每个基因的所有观察到的独特氨基酸序列变异或“等位基因”进行了识别和列举。
模型选择和训练:首先基于支持向量机(support vector machine ,SVM)方法训练模型(SVM是通过scikit-learn实现的),训练集来自六种针对金黄色葡萄球菌的抗生素治疗方法。验证是根据从文献和CARD数据库中收集的已知的的AMR基因。
然后使用相同的特征矩阵和AMR表型,对每个抗生素病例训练了两种类型的SVM合集,以将基因组分类为易感或耐药,由500个SVM组成,每个SVM训练的结果是:1)随机抽取80%的基因组和所有特征的随机样本,得到类似于中的引导合集;或2)随机抽取80%的基因组和50%的特征,得到随机子空间合集(RSE),这种调整以前被证明可以提高在高维生物数据上训练的SVM的准确性。类似地,特征按特征权重进行排序。
最后使用SVM-RSE的方法在P. aeruginosa和E.coli数据集中鉴定已知的AMR基因。