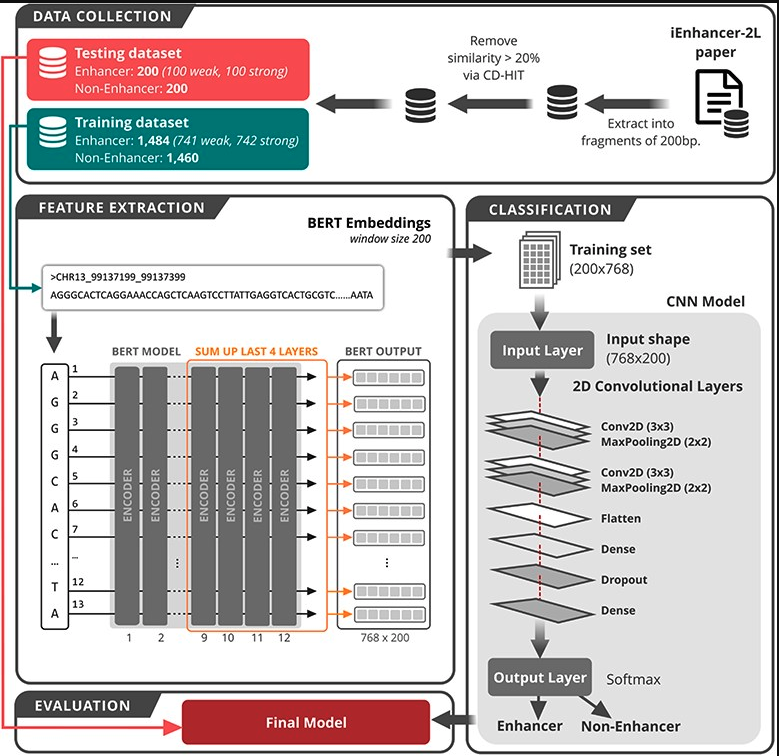
1. 基于BERT和二维卷积神经网络的transformer架构从序列信息中识别DNA enhancers
题目:A transformer architecture based on BERT and 2D convolutional neural network to identify DNA enhancers from sequence information
杂志:Briefings in Bioinformatics
IF: 13.944
时间:05 February 2021
链接:https://doi.org/10.1093/bib/bbab005
摘要
最近,语言表征模型因其显著的成果而在自然语言处理领域引起了广泛的关注。其中,bidirectional encoder representations from transformers(BERT)已被证明是一种简单而强大的语言模型,取得了新颖的最先进的性能。BERT采用了语境化词语嵌入的概念来捕捉词语的语义和出现的语境。在这项研究中,我们提出了一种新的技术,将基于BERT的多语言模型纳入生物信息学中,以表示DNA序列的信息。我们将DNA序列视为自然句子,然后使用BERT模型将其转化为固定长度的数字矩阵。作为一个案例研究,我们将我们的方法应用于DNA增强子的预测,这是该领域中一个著名的和具有挑战性的问题。然后我们观察到,与目前生物信息学中最先进的特征相比,我们基于BERT的特征在敏感性、特异性、准确性和马修斯相关系数方面提高了5-10%以上。此外,高级实验表明,深度学习(以二维卷积神经网络为代表;CNN)在学习BERT特征方面拥有比其他传统机器学习技术更好的潜力。总之,我们认为BERT和二维CNN可以在利用序列信息进行生物建模方面开辟一条新的途径。
DNA数据预处理
首先,标准FASTA格式的正常DNA序列被分割成等长的DNA片段,并选取滑动窗口的值。在我们的语言模型中,我们需要通过使用不同的n-gram级别将这些片段视为 “DNA句子”。
在这项研究中,我们通过将一个核苷酸视为自然语言中的一个词(unigram)来建立NLP模型。因此,在第二步中,每个DNA片段在碱基之间插入空格,形成一系列的核苷酸,每个核苷酸都被假装成人类语言中的一个词。
一般来说,在训练BERT模型时,研究人员经常添加特殊的标记,如CLS标记和SEP标记作为句子的开始和结束。我们假设这些标记不传达生物学意义,因此不遵循这一惯例。
2. DTSyn:一个基于双transformer的神经网络来预测协同的药物组合
题目:DTSyn: a dual-transformer-based neural network to predict synergistic drug combinations
杂志:Briefings in Bioinformatics
IF: 13.944
时间:01 August 2022
链接:https://doi.org/10.1093/bib/bbac302
摘要
药物组合疗法在许多方面都优于癌症治疗的单一疗法。由于可能的药物对的巨大搜索空间的耗时过程,通过筛选来确定新的药物组合对湿实验室来说是具有挑战性的。因此,人们开发了计算方法来预测具有潜在协同功能的药物对。尽管目前的模型很成功,但从化学-基因-组织相互作用的角度理解药物协同作用的机制缺乏研究,阻碍了目前的算法从药物机制研究。在此,我们提出了一个基于多头关注机制 (multi-head attention) 的深度神经网络模型,称为DTSyn(用于药物对协同作用预测的双transformer编码器模型),以识别新型药物组合。我们设计了一个细粒度的transformer编码器来捕捉化学亚结构-基因和基因-基因的关联,以及一个粗粒度的transformer编码器来提取化学-化学和化学-细胞系的相互作用。DTSyn达到了最高的接收操作特征曲线下面积0.73,0.78。0.82和0.81,超过了所有竞争方法。此外,DTSyn在五个独立的数据集中取得了最佳的真阳性率(TPR)。研究表明,两个transformer编码器块都对DTSyn的性能做出了贡献。此外,DTSyn可以提取化学品和细胞系之间的相互作用,代表药物作用的潜在机制。通过利用注意力机制和预训练的基因嵌入,DTSyn显示出更好的可解释性能力。因此,我们设想我们的模型是一个有价值的工具,可以通过化学品和细胞系的基因表达谱来优先考虑协同作用的药物对。
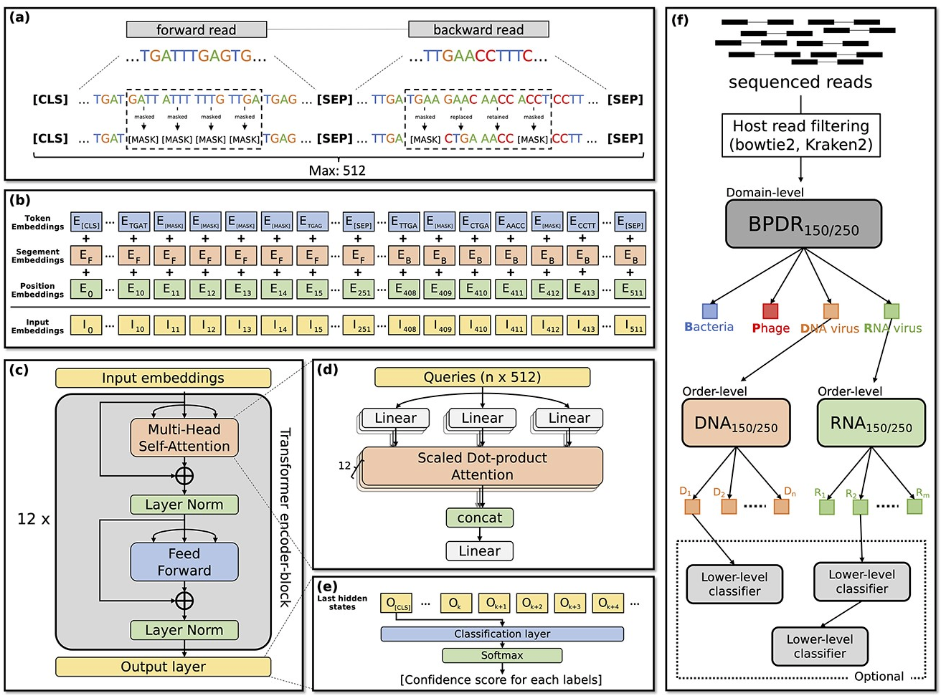
3.ViBE:利用元基因组测序数据识别真核生物病毒的层次化BERT模型
题目:ViBE: a hierarchical BERT model to identify eukaryotic viruses using metagenome sequencing data
杂志:Briefings in Bioinformatics
IF: 13.944
时间:04 June 2022
链接:https://doi.org/10.1093/bib/bbac204
摘要
病毒在人类和各种环境中无处不在,并不断地自我变异。在没有培养的环境中识别病毒是具有挑战性的;然而,促进对新型病毒的筛选和扩大对病毒空间的认识是至关重要的。基于同源性的方法,利用已知的病毒基因组识别病毒依赖于序列比对,因此很难捕捉到已知病毒的远程同源物。为了从元基因组样本中准确捕捉病毒信号,需要建立模型来理解病毒基因组的编码模式。在这项研究中,我们开发了一个层次化的BERT模型,名为ViBE,用于从元基因组测序数据中检测真核生物病毒,并对它们进行阶级分类。我们使用从病毒参考基因组产生的类读序列对ViBE进行了预训练,并得出了三个微调模型,对真核脱氧核糖核酸病毒和真核核糖核酸病毒的成对读数进行分类。ViBE取得了比最先进的基于对齐的方法更高的召回率,同时保持了相当的精度。在所有测试案例中,ViBE的表现都优于最先进的无对齐方法。ViBE的性能也用真实的测序数据集进行了验证,包括阴道病毒组。