awk是一个处理文本文件的程序,也是一门强大的编程语言。awk是把文件逐行的读入,以空格为默认分隔符将每行分割,切开的部分再进行各种分析处理,非常适用于每行格式相同的文本文件。

示例数据:
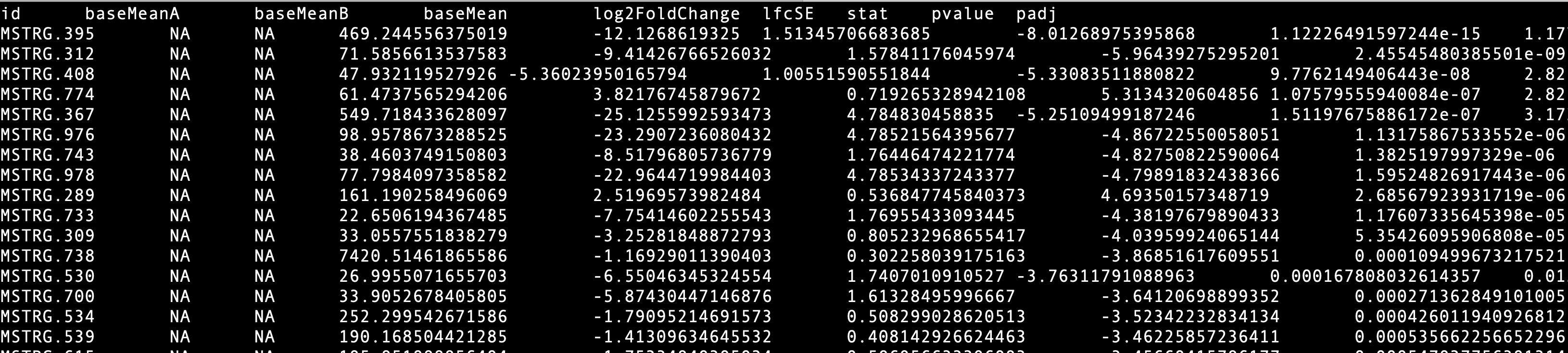
DEseq2得到的差异表达矩阵(dfdata)作为示例数据:
包括9列数据,分别为id, baseMeanA, baseMeanB,baseMean, log2FoldChange,lfcSE,stat,pvalue,padj
基本用法
- 输出文件内容
1 | # 格式 |
将输出差异基因结构的前5行
提取基因ID,log2FoldChange, pvalue三列信息
awk会根据空格和制表符等分隔符,将每一行分成若干字段,依次用$1、$2、$3代表第一个字段、第二个字段、第三个字段等,$1, $2可以理解为被分隔符分割的第一列,第二列-F:输入分隔符,这里是\t1
2# 将输出差异基因前10行的第1、5和8列,默认是以空格分割,所以输出结果
head dfdata | awk -F '\t' '{print $1,$5,$8}'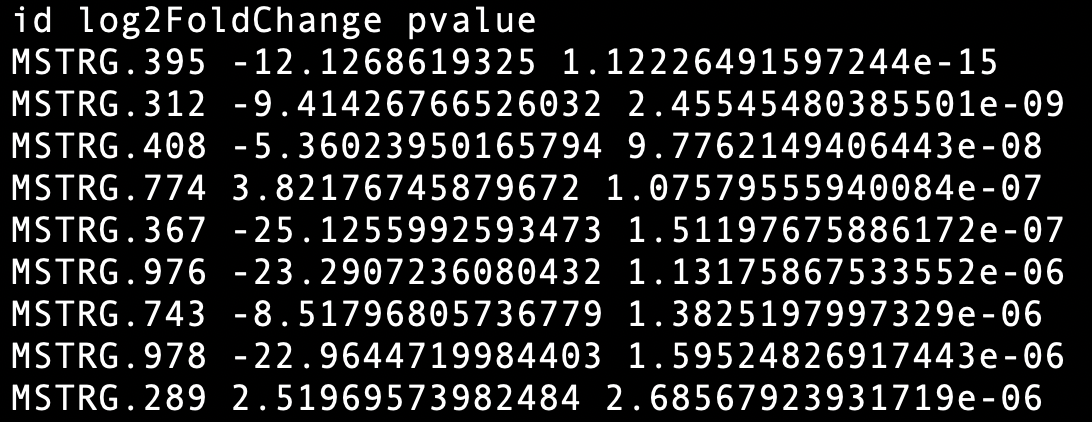
OFS: 输出分隔符1
2# 以tab分割输出
head dfdata | awk -F '\t' '{print $1,$5,$8}' OFS='\t'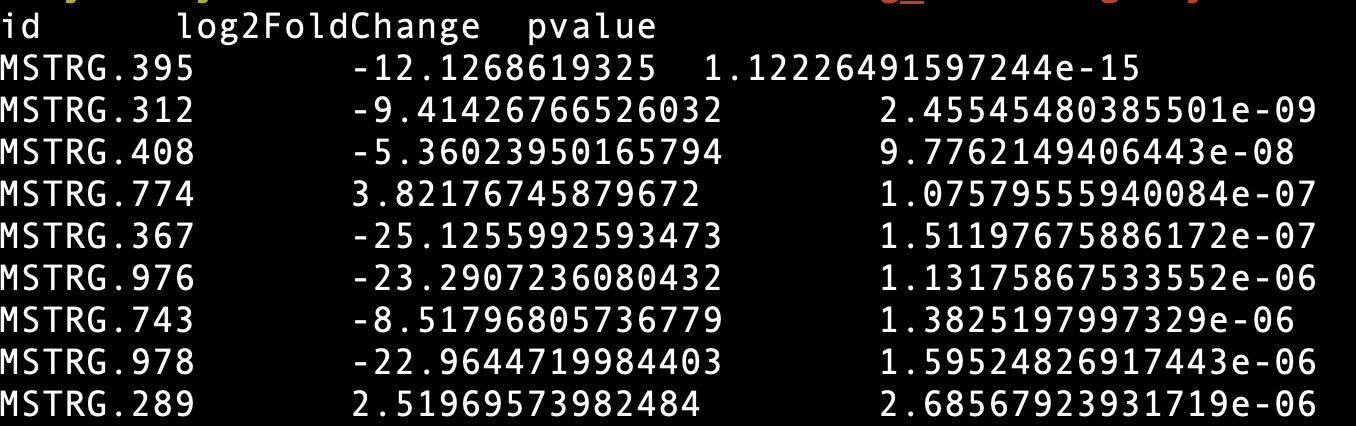
其他变量
FILENAME:当前文件名FS:字段分隔符,默认是空格和制表符。RS:行分隔符,用于分割每一行,默认是换行符。OFS:输出字段的分隔符,用于打印时分隔字段,默认为空格。ORS:输出记录的分隔符,用于打印时分隔记录,默认为换行符。OFMT:数字输出的格式,默认为%.6g
条件
1 | awk '条件 {动作}' 文件名 |
可以结合正则表达式规则使用
- 输出特定基因ID的行
1 | awk -F '\t' '/id/ {print $0}' dfdata |
if语句
判断语句和执行语句必须用在{}中,且判断语句用()扩起来
1 | awk '{if () action; else action}' file |
- 输出 |log2FoldChange | > 1且pvalue < 0.05的所有信息
符合|log2FoldChange | > 1且pvalue < 0.05的基因有多少个
1 | awk '{if ($5>1 || $5 < -1 && $8<0.05 ) print $0}' dfdata | wc |
且同时输出header
1 | awk '/id/ {print $0}; {if ($5>1 || $5 < -1 && $8<0.05 ) print $0}' dfdata | less -S |
函数
tolower():字符转为小写。length():返回字符串长度。substr():返回子字符串。sin():正弦。cos():余弦。sqrt():平方根。rand():随机数
1 | awk '/id/ {print $0} dfdata |